BETA THALASSAEMIA
what is beta thallasemia?
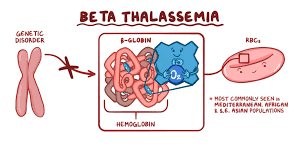
People do not produce enough beta protein have beta Thalassaemia. It is found in people of Mediterranean descent such as Italians and Greeks, and is also found in the Arabian Peninsula, Iran, Africa, Southeast Asia and Southern china.
Beta globin is made by two genes, one on each chromosome 11. Individuals who have one abnormal beta globin gene have beta Thalassaemia trait.
BETA THALASSAEMIA MINOR or BETA THALASSAEMIA TRAIT
In beta Thalassaemia trait, one of the two genes is abnormal but the lack of beta protein is not great enough to cause problems in the normal functioning of the hemoglobin. A person with this condition simply carries the genetic trait for beta thalassemia and will usually experience no health problems other than a mild anemia.
As in alpha thalassemia minor, physicians often mistake the small red blood cells of the person with beta thalassemia minor as a sign of iron- deficiency anemia and incorrectly prescribe iron supplements.
Here are key points about beta thalassemia:
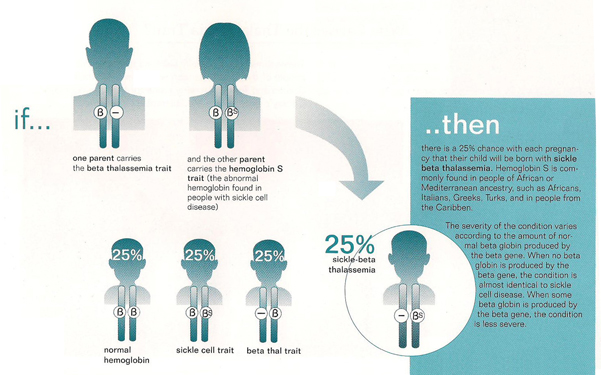
Genetic Inheritance: Beta thalassemia is inherited in an autosomal recessive pattern. This means that a person must inherit two mutated genes (one from each parent) to develop the disorder. If a person inherits only one mutated gene, they are carriers (beta thalassemia trait) and typically do not have symptoms.
Types and Severity:
- Beta Thalassemia Minor (Trait): Individuals have one mutated beta globin gene and one normal gene. They are carriers and usually have mild or no symptoms of anemia. Treatment is generally not needed.
- Beta Thalassemia Intermedia: Individuals have two mutated beta globin genes. Symptoms are more severe than in beta thalassemia minor but less severe than in beta thalassemia major.
- Beta Thalassemia Major (Cooley’s Anemia): Individuals have two mutated beta globin genes, leading to severe anemia that requires regular blood transfusions from early childhood. Without treatment, beta thalassemia major can be life-threatening due to complications from severe anemia.
Symptoms:
- Beta Thalassemia Minor: Mild anemia, fatigue, and possibly slight jaundice.
- Beta Thalassemia Intermedia: Moderate to severe anemia, growth problems, bone abnormalities, enlarged spleen and liver (hepatosplenomegaly), and possible facial bone deformities.
- Beta Thalassemia Major: Severe anemia, failure to thrive in infancy, pale or jaundiced skin, bone deformities, enlarged spleen and liver, and complications such as heart problems and infections.
Diagnosis: Diagnosis is typically made through blood tests that measure hemoglobin levels and identify abnormal hemoglobin variants. Genetic testing can confirm the presence of mutated beta globin genes.
Treatment:
- Blood Transfusions: Regular transfusions are necessary for individuals with beta thalassemia major to maintain adequate hemoglobin levels and prevent severe anemia.
- Iron Chelation Therapy: Since frequent transfusions can lead to iron overload, chelation therapy is used to remove excess iron from the body.
- Bone Marrow Transplantation: This may be curative for some individuals with beta thalassemia major, providing a healthy source of hematopoietic stem cells that can produce normal hemoglobin.
Prognosis: The prognosis varies depending on the type and severity of beta thalassemia. With regular transfusions and appropriate medical care, individuals with beta thalassemia major can live into adulthood. Advances in treatment, including bone marrow transplantation, have improved outcomes for many patients.


The Parents’ Association Thalassemic is the national non –profit health organization dedicated to serving patients afflicted with various forms of Thalassaemia most notably the major forms of this generic blood disease